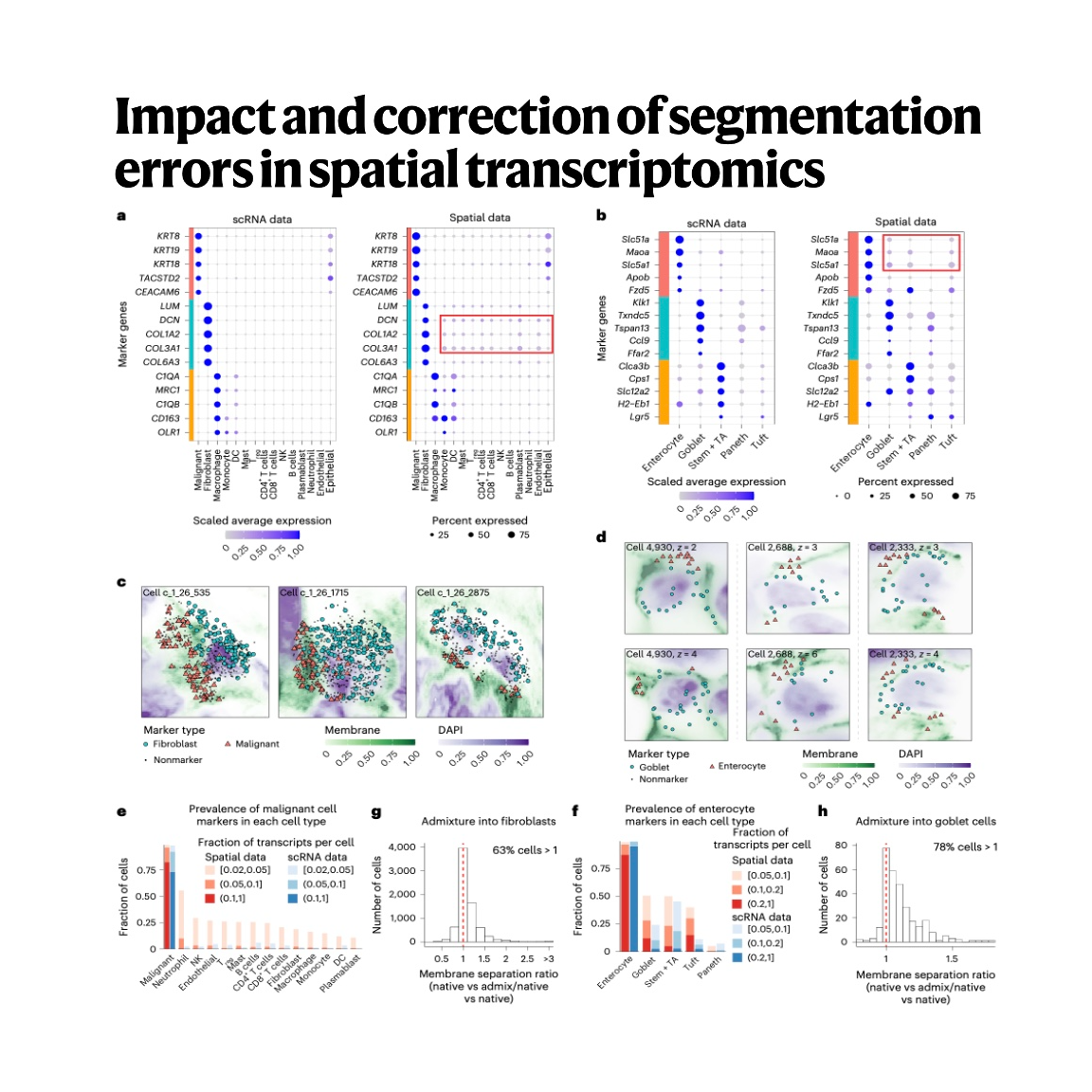
Spatial transcriptomics relies on sequencing and imaging to map cellular activities, but both methods face significant technical hurdles in accurately assigning RNA molecules to individual cells. The provided text highlights how cell segmentation errors and the limitations of two-dimensional mapping lead to "admixture," where transcripts from neighboring cells are incorrectly attributed to a speci...去小宇宙查看完整单集简介
前往小宇宙评论区与主播互动